作者简介:顾建文,著名脑外科专家,博士导师,解放军第306医院院长,主任医师,全军神经外科副主委,中华医学会理事。
苯丙酮尿症(phenylketonuria,PKU)或称苯丙氨酸羟化酶缺乏症(phenylalanine hydroxylase deficiency)。PKU是氨基酸代谢性疾病最常见的类型,全球发病率约1/1.5万,随民族和地区而不同。与其他氨基酸尿症不同,该病有特殊的历史意义。
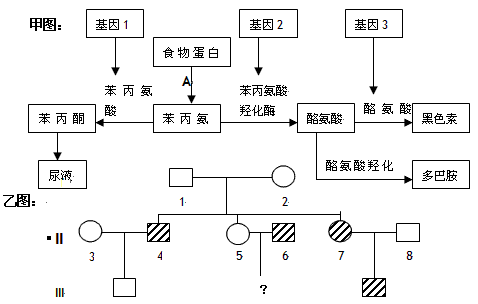
丙氨酸(phenylalamine,PA)是一种人体必需氨基酸,它参与构成各种蛋白质成分,但在人体内不能合成。正常情况下,摄入的PA中约有50%左右用于合成各种成分的蛋白质,其余部分在苯丙氨酸羟化酶的作用下变为酪氨酸,再经其他酶的作用转化为多巴、多巴胺、肾上腺素、去甲肾上腺素及黑色素等。苯丙氨酸羟化酶是复合酶系统,除羟化酶本身外,还包括二氢蝶呤还原酶及辅酶四氢生物蝶呤,任何一种酶缺陷均可引起血苯丙氨酸增高。当PA羟化酶缺乏时,未参与第一步蛋白质合成的苯丙氨酸在血浆内贮积,并沉积于全身组织包括脑。血中苯丙氨酸超过肾阈而排出,产生苯丙氨基酸尿。
PA的主要途径(羟化)受阻后,PA的次要代谢途径则代偿性地亢进,使PA转化为苯丙酮酸、苯乳酸、正羟苯乙酸和苯乙酸的比重逐步增加。正常时这条代谢旁路进行得很少,因此这些代谢产物的含量极少;当PA羟化酶缺乏时,这些代谢产物才达到异常增高的水平,积蓄于组织、血浆和脑脊液中,并大量从尿中排出,产生苯丙酮尿症。
1.典型PKU病例出生时多表现正常,在1~6个月后婴儿逐步出现智商(IQ)降低,并出现易激惹,呕吐,过度活动或焦躁不安,有些婴儿出现湿疹。身体或衣服可闻到特殊的气味,如霉味或“鼠味”,是该病患儿的特征性表现。智力低下是本病最常见的症状,约90%以上的患儿可有中至重度智力低下,6个月以后IQ迅速下降,至1岁时降至50;3岁时降到40左右; 5~6岁时测定IQ评分通常<20,偶尔为20~50,很少>50。

2.患儿1岁后运动发育也明显落后,语言障碍最突出,可有步态笨拙、双手细震颤、协调障碍、姿势怪异及重复性手指作态等。行为异常表现为多动、易激惹、激越行为和情绪不稳等,见于约60%以上的患儿。
3.癫痫发作是本病的又一特征,常在1岁左右发病,约25%的严重智力迟钝患儿可有癫痫发作。临床表现最常见为屈肌痉挛(flexor spasm),其次为失神性发作和全面性强直-阵挛性发作,也可见婴儿痉挛症。随年龄长大,婴儿痉挛发作减少,转变为小发作或大发作。
4.神经系统体格检查异常发现不多,1/3患儿正常;1/3有轻微多动、震颤、腱反射亢进、踝阵挛等;锥体束征较常见;不自主运动如扭转痉挛、手足徐动、肌张力障碍等以及明显小脑性共济失调也有过报道,但很少见。严重者可出现脑性瘫痪。
5.一般体态、生长发育多数正常;90%的患儿有黑色素缺乏,皮肤特别白,但又不是白化病,头发淡黄或棕色,虹膜色素淡呈棕黄色,白种人呈蓝色。此外,小孩的前牙稀疏、骈指、脊柱裂等亦可常见。
6.脑电图检查80%可见异常。CT可见脑萎缩。
苯丙酮尿症的症状和体征除智能低下外大部分是可逆的。当PA浓度控制后症状可以消失,癫痫可以控制,脑电图异常可以恢复,毛发色素可以加深,身体气味可以消失。
治疗目的在于减少体液中苯丙氨酸及代谢产物含量,防止或减轻脑损害。

1.典型PKU的治疗关键是控制饮食中苯丙氨酸(PA)的含量,采取低苯丙氨酸饮食,婴儿期可用人工合成低苯丙氨酸奶粉喂养,国内也有出品,既能满足机体代谢和生长发育的最低需要,又不会使血中 PA含量过高造成脑损伤。饮食控制要及早开始,如生后6个月开始疗效差,4~5岁开始因神经系统发育已基本完成而无效(Holtzman etal,1986)。如控制患儿饮食中PA,血PA含量维持在0.18~0.92mmol/L(5~10mg/dl)疗效可能较理想,可提高智力发育。然而,断奶后再维持低苯丙氨酸饮食并非容易,因动植物蛋白中PA含量均可达0.18~0.31mmol/L,必须严格限制蛋白质饮食,还要充分满足热卡、脂肪、维生素及矿物质等需求,需要营养医师制定食谱,进行严密随访。特殊膳食的口味很差,许多患儿难于接受。文献报告饮食控制愈好,IQ发育愈高。如儿童期中断饮食治疗,可再次出现精神运动发育迟滞,IQ下降;恢复饮食治疗,仍可望获得一定疗效。国内许多病例是在婴儿后期或儿童期已发生脑损伤时才确诊,此时采取饮食治疗虽不能改善智商,但对控制痫性发作及改善发育迟滞、皮肤湿疹、皮肤及毛发色素减退、硬皮症、精液减少和生殖功能缺陷等仍有裨益。
目前,饮食治疗的期限仍有争论。多数患儿6岁后可不必严格限制膳食,但苯丙氨酸摄入仍在限制之列。有苯丙氨酸血症而无PKU的儿童无必要饮食治疗。也有学者认为,饮食治疗应持续至青春期后以至终生。对饮食治疗的副作用,如生长落后、短身材、低体重、低血糖、低蛋白血症及骨龄延迟等必须有充分预料。因PA是人体必需氨基酸,完全缺乏可能导致严重后果,如嗜睡、贫血、厌食、腹泻和皮疹,甚至死亡。
2.少数患儿是PKU变异型,限制PA饮食不能阻止神经系统受累,有些这类患儿早在新生儿早期即有肌张力障碍性锥体外系肌强直,称僵婴综合征(stiff-baby syndrome),用生蝶呤(生物蝶呤)可能有效,因这类患儿肝脏苯丙氨酸羟化酶水平正常,该酶缺乏是由于四氢生物蝶呤活性协同因子合成障碍,可因二氢蝶呤降解酶缺乏或生蝶呤(生物蝶呤)合成减少所致;尿中儿茶酚胺代谢物和5-羟色胺减少,限制饮食PA无效,对这些病例的治疗主要通过补充神经递质前体左旋多巴(L-dopa)和5-羟色氨酸(5-hydroxytryptophan)加以纠正。
饮食保健

不要吃肥肉,饮食中适量控制糖份。
遗传病治疗困难,疗效不满意,预防显得更为重要。预防措施包括避免近亲结婚,推行遗传咨询、携带者基因检测及产前诊断和选择性人工流产等,防止患儿出生。
提倡母乳喂养。尽早发现苯丙酮尿症携带者和普及三氯化铁尿布,使已致病婴儿及早发现、及早治疗是预防智能低下的重要办法。
随着年龄的增大,摄入的苯丙氨酸用于合成蛋白的量逐渐减少。出生以后,每天摄入的苯丙氨酸约为0.5g,儿童和成人增加到4g。其中较大部分被氧化成酪氨酸,这一过程主要依赖于苯丙氨酸羟化酶(PAH),但也需要辅因子参与。如果这一氧化过程发生障碍,则有苯丙氨酸在体内堆积,在此情况下,苯丙氨酸则通过其他途径进行代谢而产生苯丙酮酸有害物质。苯丙酮尿(PKU)就是因为PAH活性减低或缺如而引起的一种遗传性疾病。PAH活性减低还可使酪氨酸受抑而使黑色素生成减少,羟苯丙酮酸酶受抑而使羟苯酮酸在体内堆积。
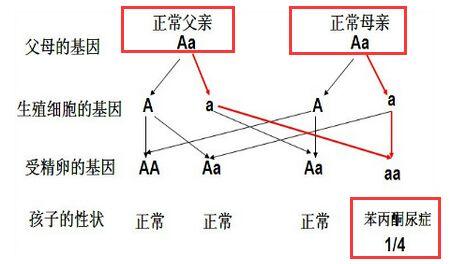
本病为常染色体隐性遗传,突变基因位于12号染色体长臂(12q24.1),该基因的微小变异即可引起发病,并非由于基因缺失。系由两个杂合子的婚配而导致的遗传性疾病,以近亲结婚的子代为多见,患儿同胞约40%患病。由于苯丙氨酸羟化酶(phenylalanine hydroxylase)基因突变,导致肝脏中苯丙氨酸羟化酶缺乏,是本病基本的生化异常。如果发生变异的碱基对不同,引起临床表现的严重程度有很大差异,可表现为典型PKU或轻度高苯丙氨酸血症。
需与其他病因(如脂类沉积病、围生期疾病)导致的精神发育迟缓,癫痫发作、震颤、肌张力增高、共济失调、腱反射亢进及脑性瘫痪相鉴别。

实验室检查:
1.尿苯丙酮酸试验由于患儿尿中排出苯丙酮酸增多,可作定性试验。方法有:
(1)三氯化铁试验:将5%三氯化铁滴入5ml尿中立即出现绿色反应则为阳性。新生儿尚未喂食,此试验呈阴性。枫糖尿病者尿也可呈阳性,故此试验特异性较差。
(2)2,4-硝基苯肼试验:如果产生黄色混浊沉淀则为阳性。
2.血苯丙氨酸测定正常人血中苯丙氨酸为60~180μmol/L,PKU患者可高达600~3600μmol/L。如果以258μmol/L为正常人与PKU病人的分界点,则有高达4%的假阳性。用色层析法则在生后几天的新生儿中可出现假阴性。MS/MS法可减少假阳性率,此方法可同时测定血苯丙氨酸和酪氨酸,并可计算苯丙氨酸/酷氨酸比值。如果以比值2.5为正常儿童与患PKU者的分界点,则可将假阳性减少到1%。故目前多用此方法来筛选新生儿苯丙酮尿症。此方法还可用来筛选半乳糖血症、枫糖尿病、同型胱氨酸尿症和先天性甲状腺功能减低症,一次检查可以筛选多种先天性疾病。
其他辅助检查:
1.脑电图(EEG):主要是棘慢波,偶见高波幅节律紊乱。EEG随访研究显示,随年龄增长,EEG异常表现逐渐增多,至12岁后EEG异常才逐渐减少。
2.产前检查:由于绒毛及羊水细胞测不出苯丙氨酸羟化酶活性,所以产前诊断问题长期不能解决。目前我国已鉴定出25种中国人PKU致病基因突变型,约占我国苯丙氨酸羟化酶突变基因的80%,已成功用于PKU患者家系突变检测和产前诊断。
3.X线检查:可见小头畸形,CT和MRI可发现弥漫性脑皮质萎缩等非特异性改变。并发症约2/3的患儿有轻度小颅畸形,眼底正常,无内脏肿大或骨骼异常。
本病的预后取决于PAH活性减低的程度和是否能早期诊断和早期治疗。轻度PAH活性减低者,可能只有轻度高苯丙氨酸血症,智力不受损害,能够得到早期诊断和早期治疗者预后良好。
1.根据生化缺陷的不同可分为
(1)典型PKU:先天性苯丙氨酸羟化酶缺陷。
(2)持续性高苯丙氨酸血症:见于苯丙氨酸羟化酶异构酶缺陷或典型苯丙酮尿症杂合子,血苯丙氨酸增高。
(3)一过性轻度高苯丙氨酸血症:多见于早产儿,是苯丙氨酸羟化酶成熟延迟所致。
(4)苯丙氨酸转氨酶缺乏:虽然血苯丙氨酸含量增加,但尿中苯丙酮酸及羟苯乙酸可不增高,给予负荷量的苯丙氨酸口服后血酪氨酸也不增加。
(5)二氢蝶呤还原酶缺乏:酶活性完全或部分缺乏,除影响脑发育外可使基底节钙化。
(6)二氢蝶呤合成缺陷:缺乏甲醇氨脱水酶或其他多种酶。
典型PKU患儿出生时神经系统正常,由于纯合子患儿缺乏神经系统保护措施,神经系统长期暴露于苯丙氨酸而出现症状。如母亲是纯合子,血苯丙氨酸水平很高,患儿是杂合子,在子宫中就可发生中枢神经系统损害,出生时表现为智力障碍。
普通型PKU及某些轻度和严重变异型,疾病早期未经治疗可出现精神衰退。推测可能为等位基因突变型,表现为高苯丙氨酸血症(hyperphenylalaninemia),无苯丙酮尿症及神经系统受累。此外,即使少数(约3%)病人控制高苯丙氨酸血症,也不能预防神经系统病变的进展。